7. Read alignment track (-bamplot read)¶
7.1. Layout options¶
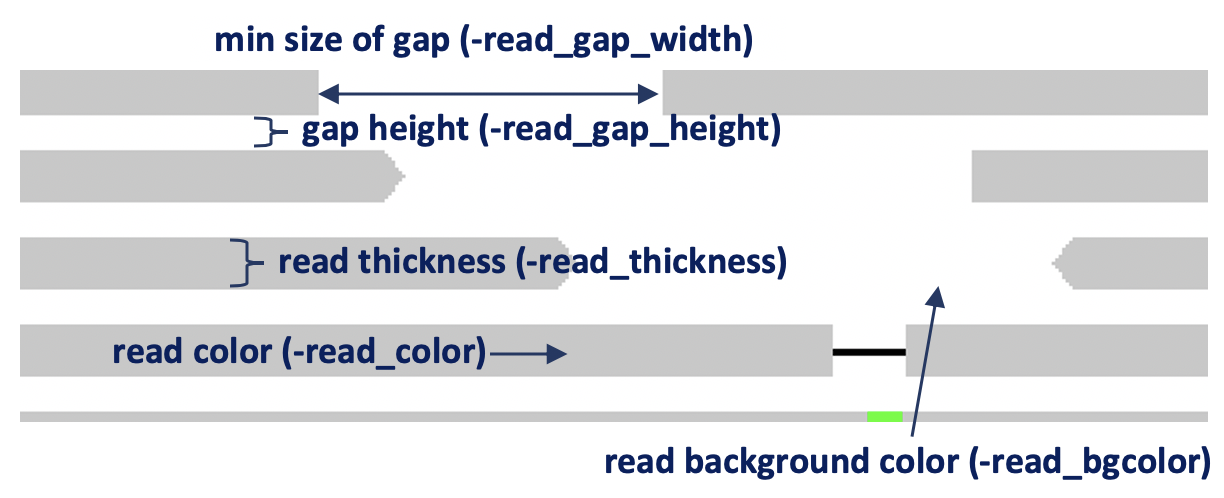
-read_thickness(default=5) : read thickness (unit:px)-read_gap_height(default=2) : read gap height (unit:px)-read_gap_width(default=2) : min size of read gap width (unit:px)-read_bgcolor(default=’FFFFFF’) : read background color-read_color(default=’C8C8C8’) : read color-center_line(default=false): draw center line-no_target_line(default=false): do not draw target line
7.2. Read group (-read_group)¶
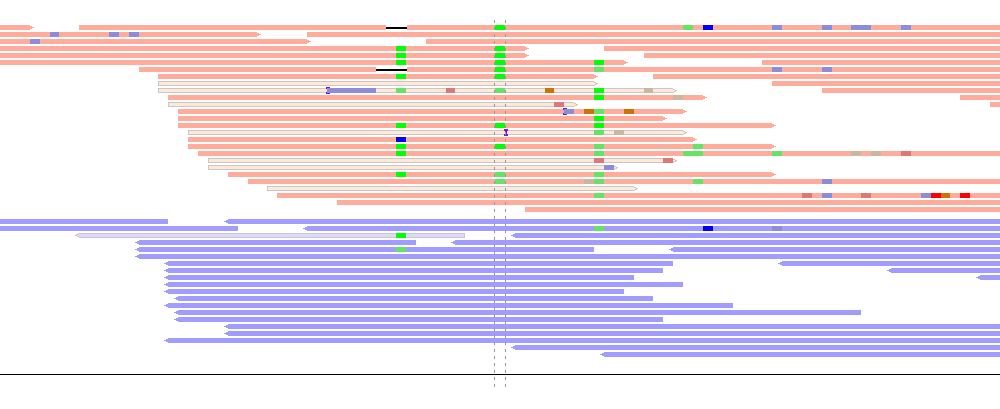
It is possible to plot the reads grouped by strand using the -read_group strand option.
1 2 3 4 5 6 7 8 | $ bamsnap \
-bam ./data/NA12879.bam \
-pos chr10:117542948 \
-no_title \
-draw bamplot \
-bamplot read \
-out ./out/NATRIO_chr10_117542948_6.png \
-read_group strand
|
7.3. Read color (-read_color_by)¶
The program provides color sets for strand and chromosomes.
7.3.1. Color by strand (-read_color_by strand)¶
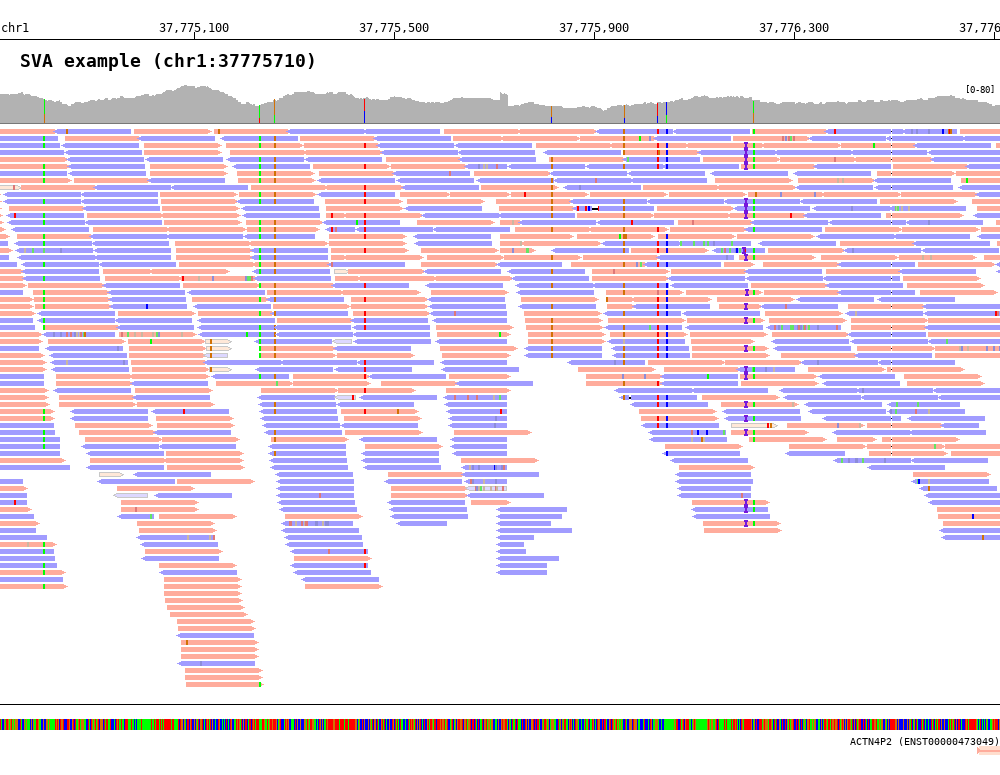
1 2 3 4 5 6 7 8 9 10 | $ bamsnap \
-bam ./data/test_SV1_chr1_37775710.bam \
-title "SVA example (chr1:37775710)" \
-pos chr1:37775710 \
-out ./out/test_SV1-4.png \
-bamplot coverage read \
-margin 1000 \
-no_target_line \
-read_color_by strand \
-save_image_only
|
The reads color by strand can be defined using -read_pos_color and -read_neg_color options.
-read_pos_color(default=’FFAC9C’) : positive strand read color-read_neg_color(default=’A19CFF’) : negative strand read color
7.3.2. Color by inter-chromosomal rearrangements (-read_color_by interchrom)¶
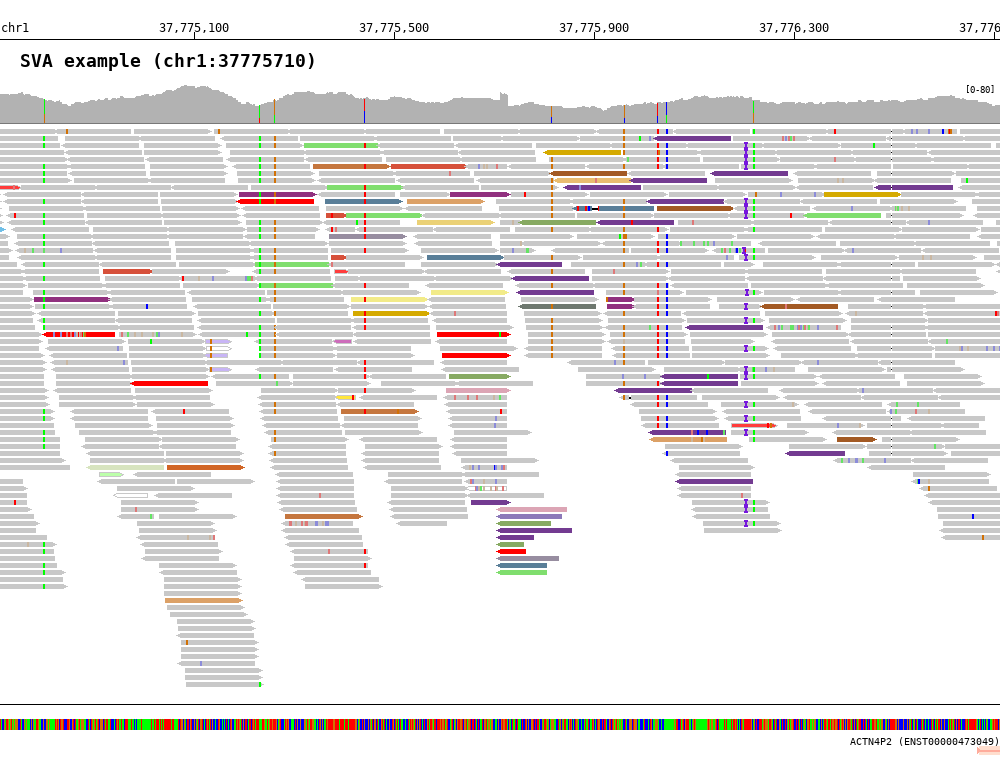
1 2 3 4 5 6 7 8 9 10 | $ bamsnap \
-bam ./data/test_SV1_chr1_37775710.bam \
-title "SVA example (chr1:37775710)" \
-pos chr1:37775710 \
-out ./out/test_SV1-3.png \
-bamplot coverage read \
-margin 1000 \
-no_target_line \
-read_color_by interchrom \
-save_image_only
|
The reads color by chromosome can be defined using -read_color_interchrom_chr1, -read_color_interchrom_chr2, .., and -read_color_interchrom_chrY options.
- Default color codes
- chr1: #64689b ⬅⬅⬅⬅
- chr2: #D6503A ⬅⬅⬅⬅
- chr3: #87AA62 ⬅⬅⬅⬅
- chr4: #F2EB89 ⬅⬅⬅⬅
- chr5: #597E98 ⬅⬅⬅⬅
- chr6: #C5763E ⬅⬅⬅⬅
- chr7: #70BFE7 ⬅⬅⬅⬅
- chr8: #91307F ⬅⬅⬅⬅
- chr9: #80DE6E ⬅⬅⬅⬅
- chr10: #DCA5B5 ⬅⬅⬅⬅
- chr11: #A35A24 ⬅⬅⬅⬅
- chr12: #978DA0 ⬅⬅⬅⬅
- chr13: #D16525 ⬅⬅⬅⬅
- chr14: #DCA167 ⬅⬅⬅⬅
- chr15: #8C79B9 ⬅⬅⬅⬅
- chr16: #E9BD71 ⬅⬅⬅⬅
- chr17: #4B2669 ⬅⬅⬅⬅
- chr18: #D7E4BF ⬅⬅⬅⬅
- chr19: #733B91 ⬅⬅⬅⬅
- chr20: #BC2D7A ⬅⬅⬅⬅
- chr21: #EBD176 ⬅⬅⬅⬅
- chr22: #6E786F ⬅⬅⬅⬅
- chrX: #D5AA00 ⬅⬅⬅⬅
- chrY: #A9D400 ⬅⬅⬅⬅
- other chromosome: #555555 ⬅⬅⬅⬅
7.4. Show soft clipped part (-show_soft_clipped)¶
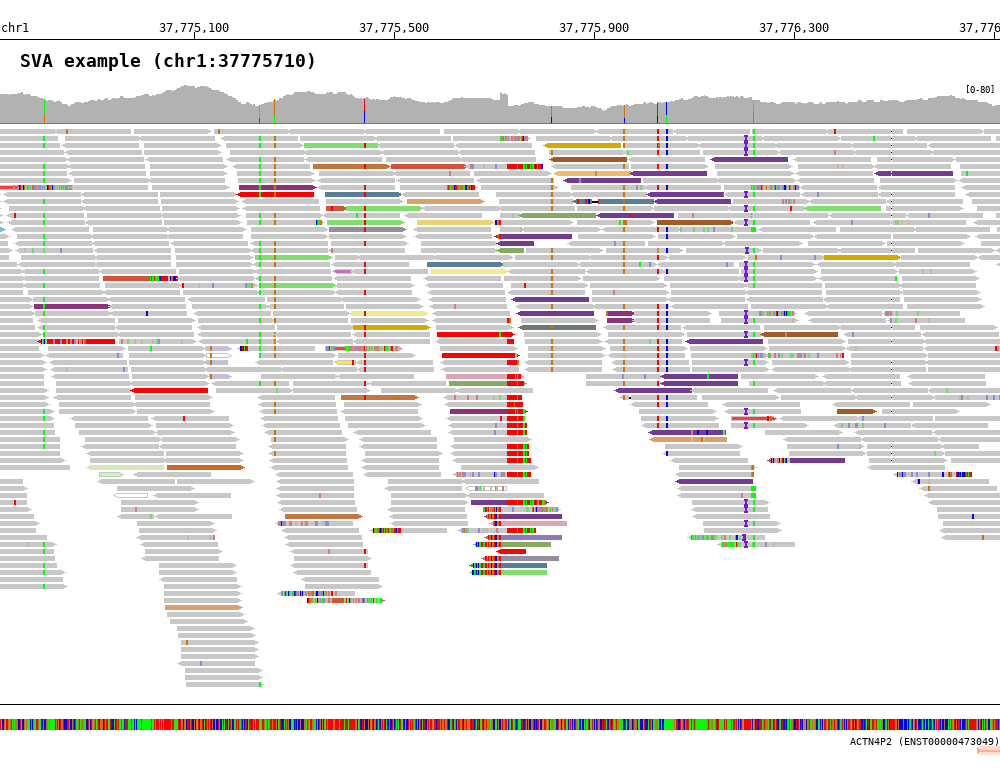
1 2 3 4 5 6 7 8 9 10 11 | $ bamsnap \
-bam ./data/test_SV1_chr1_37775710.bam \
-title "SVA example (chr1:37775710)" \
-pos chr1:37775710 \
-out ./out/test_SV1-3_1.png \
-bamplot coverage read \
-margin 1000 \
-no_target_line \
-show_soft_clipped \
-read_color_by interchrom \
-save_image_only
|
7.5. Deletion¶
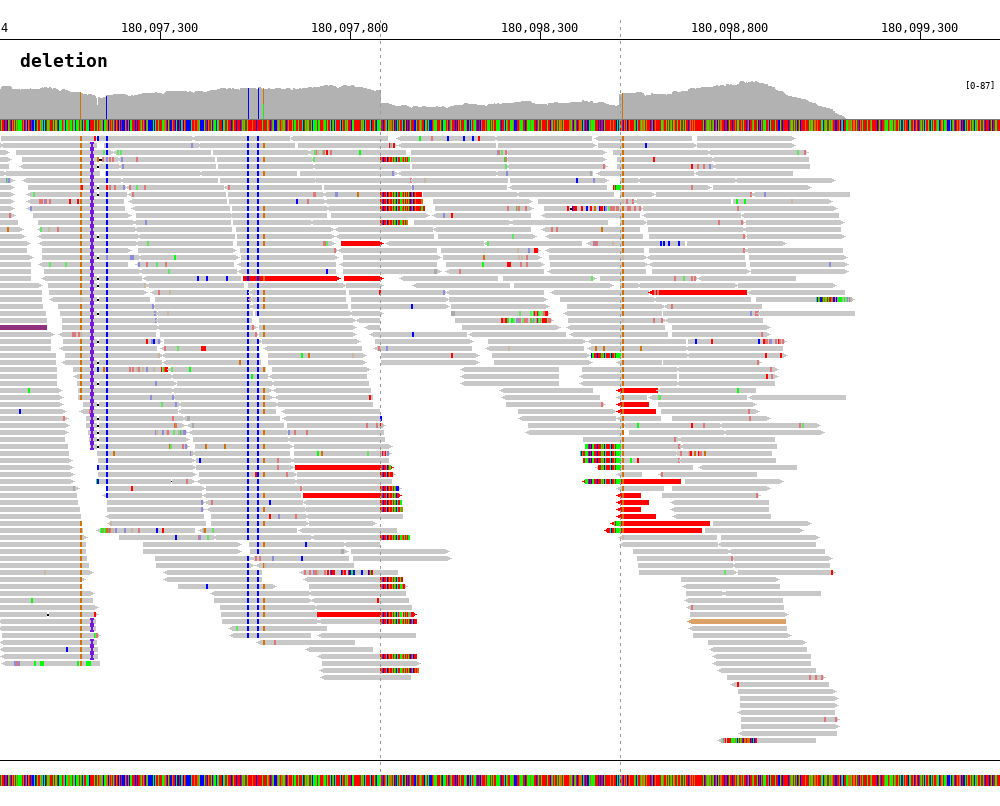
1 2 3 4 5 6 7 8 9 10 | $ bamsnap \
-bam ./data/test_DEL_4_180097876_180097877.bam \
-pos 4:180097878-180098507 \
-margin 1000 \
-title deletion \
-out ./out/test_DEL_1.png \
-refversion hg19 \
-show_soft_clipped \
-read_color_by interchrom \
-save_image_only
|
The insert size threshold between read mates to detect deletions is set by -insert_size_del_threshold (default is 1000).
The color of reads for deletion is #FF0000 by default. You can change the color using -read_color_deletion option.